
The results of the Taxon dataset are presented in details here.
The main focus of the evaluation here is to be task-oriented. First, we manually evaluate the quality of the generated alignments, in terms of precision. Second, we evaluate the generated correspondences using a SPARQL query rewriting system and manually measure their ability of answering a set of queries over each dataset.
We excluded from the evaluation the generated correspondences implying identical IRIs such as owl:Class = owl:Class. As instance matching is not the focus of this evaluation, we do not consider instance correspondences. In order to measure the precision of the output alignments, each correspondence (except if in the categories mentioned above) was manually verified and classified as true positive or false positive. A true positive correspondence has semantically equivalent members. All other correspondences are false positives. The confidence of the correspondence is not taken into account. All the systems output only equivalence correspondences so the subsumption relation was not considered.
We give two precision measures:
The queries written for the source ontology were rewritten: automatically when dealing with (1:1) or (1:n) correspondences using the system described in [1], and manually when dealing with (m:n) correspondences. The alignment used for the rewriting process is the union of the source-target and target-source alignment to take into account oriented genereated alignments. If a source member included in the query is the left member of many correspondences, a target query is created for each of these correspondences. A source query is considered successfully rewritten if at least one of the target queries is semantically equivalent.
On the 3 systems applicated to the Complex track, AMLC needed an input simple alignment so could not be run on the Taxon track. XMap encountered an error in the execution and withdrew its application. We ran all the systems which applied for the Complex, Conference and Anatomy tracks: ALOD2Vec, AML, AMLC, CANARD, DOME, FCAMapX, Holontology, KEPLER, Lily, LogMap, LogMapBio, LogMapLt, POMAP++, XMap. Out of the 14 tested systems, 7 did not output any alignment or crashed:
All the other systems (AML, CANARD, LogMap, LogMapBio, LogMapLt, POMAP++) output at least an alignment. AML only output one alignment as it encountered errors during the loading phase of AgronomicTaxon and DBpedia. The only system to output complex correspondences was CANARD.
The systems have been executed on a Ubuntu 16.04 machine configured with 16GB of RAM running under a i7-4790K CPU 4.00GHz x 8 processors. All measurements are based on a single run.
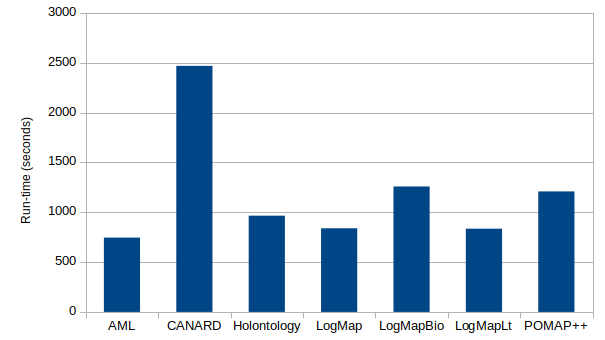
The following table presents the results of the systems on the Taxon track. The table presents the run-time, the number of output correspondences, evaluated correspondence (number of output correspondences minus same IRI or instance correspondences), the number of correct correspondences. The global precision is a macro average of the precision (number of correct correspondences on all pairs divided by the number of evaluated correspondences on all pairs). The average precision is the micro average of the precision for each pair of ontologies. The number of (1:1), (1:n) and (m:n) output correspondences is also reported. Finally, the qwr is the percentage of queries well rewritten using the output alignments.
| System | Runtime (s) | output corres. | eval. corres. | correct corres. | Global Precision | Average Precision | (1:1) | (1:n) | (m:n) | QWR |
|---|---|---|---|---|---|---|---|---|---|---|
| AML | 745 | 4 | 1 | 0 | 0.00 | 0.00 | 4 | 0 | 0 | 0.00 |
| CANARD | 2468 | 142 | 142 | 27 | 0.19 | 0.20 | 4 | 66 | 72 | 0.13 |
| Holontology | 965 | 44 | 13 | 3 | 0.23 | 0.22 | 44 | 0 | 0 | 0.00 |
| LogMap | 839 | 48 | 19 | 10 | 0.53 | 0.54 | 48 | 0 | 0 | 0.07 |
| LogMapBio | 1258 | 45 | 17 | 5 | 0.29 | 0.28 | 45 | 0 | 0 | 0.00 |
| LogMapLt | 834 | 5064 | 1920 | 10 | 0.01 | 0.16 | 5064 | 0 | 0 | 0.10 |
| POMAP++ | 1208 | 33 | 8 | 2 | 0.25 | 0.14 | 33 | 0 | 0 | 0.00 |
CANARD was the only system able to output complex correspondences on this track. LogMapLt generated the highest number of correspondences, but most were incorrect (global precision of 0.01). Overall, the best global precision is obtained by LogMap, followed by LogMapBio and POMAP++. In terms of average precision, LogMap is followed by LogMapBio and Hontology. However, these systems are not able to generate any complex correspondences.
Overall, the query cases needing simple alignments were rather well covered by simple matchers: 42% (4+2/6+8) of them for LogMapLt, 60% (4+2/4+6) if we consider only simple equivalence and leave out the queries needing instance matching; 29% and 40% for LogMap. For the query cases needing complex correspondences, (0+6/28+16) 14% were covered by CANARD. For all the query cases, the CANARD system could provide an answer to 8 query cases out of the 36 + 24 = 60 cases.
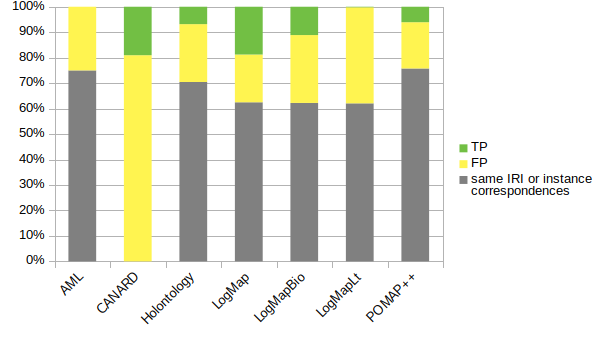
The next figure shows the proportion of correspondences output by the systems: not considered in the evaluation, incorrect (FP) and correct (TP). All the systems except CANARD output over 60% of of same IRIs or instance correspondences.
Looking to the ability of rewriting queries using the generated alignments, simple matchers could cover the most cases involving such simple correspondences. However, the main take-away of this first evaluation is that simple alignments are sometimes not sufficient for some applications. Only the CANARD system could output complex correspondences but limited to the unary queries.
With respect to the technical environment, the SEALS system as it is now was probably not adapted to deal with big knowledge bases as the loading phase got very slow. The use of SPARQL endpoints instead of two ontology files would make more sense given that many LOD repositories provide one. As the evaluation was based on a set of queries, the systems could receive the queries as input in order to limit the search space and therefore gain in efficiency.
[1] Thieblin, E., Amarger, F., Haemmerle, O., Hernandez, N., Trojahn, C.: Rewriting SELECT SPARQL queries from 1:n complex correspondences. In: 11th ISWC workshop on ontology matching. pp. 49–60 (2016)